03 Analysis
Every run is fully reproducible — trajectories analyzed with MDTraj, figures generated from the raw output.
A full OpenMM pipeline stood up on a single workstation GPU: install → simulate → analyze → visualize. Explicit- and implicit-solvent runs of small systems, with GPU-vs-CPU timing. 在一張工作站顯卡上跑通的完整分子動力學流程 — 從環境安裝到模擬、分析、視覺化。給合作者看我們做了哪些模擬與效能。
Three representative systems spanning explicit solvent (full water + PME) and implicit solvent (Generalized Born). All run on the CUDA platform with a Langevin Middle integrator, 2 fs timestep, 300 K.
| System | Solvent | Atoms | Force field | Length | Wall time | Throughput |
|---|---|---|---|---|---|---|
| TIP3P water box 2 nm cube |
explicit · PME | 774 | AMBER14 / TIP3P | 10 ps | 1.5 s | 633 ns/day |
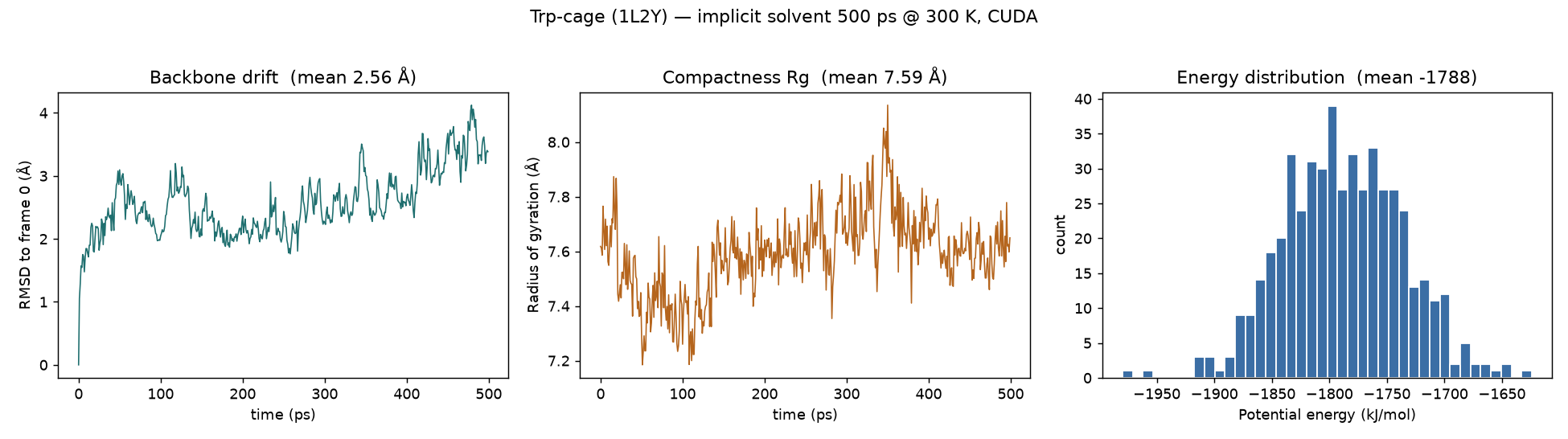
| Trp-cage (TC5b) PDB 1L2Y · 20 res |
implicit · GBn2 | 304 | ff14SB + GBn2 | 500 ps | 44.7 s | 967 ns/day |
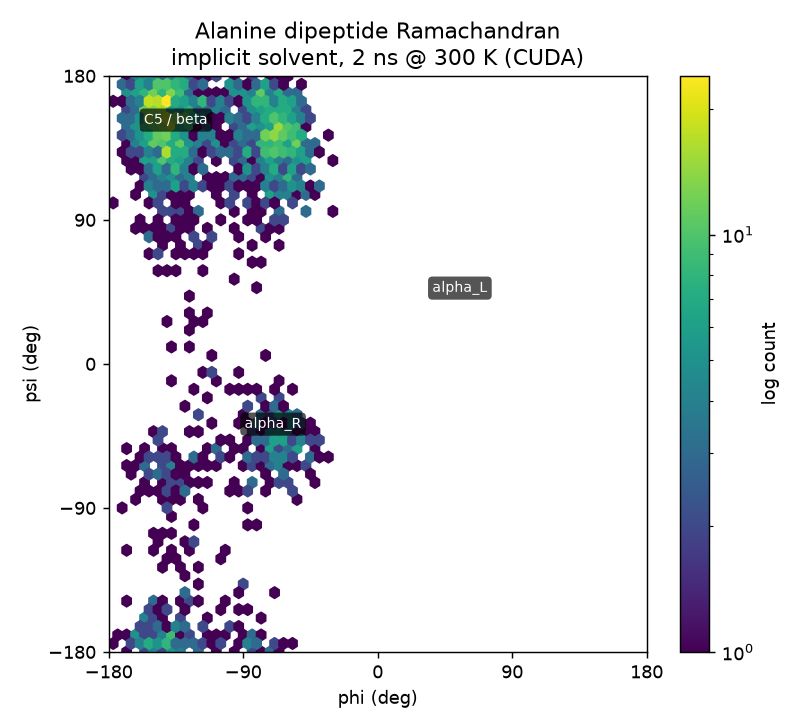
| Alanine dipeptide Ace-Ala-Nme |
implicit · OBC | 22 | AMBER / OBC | 2 ns | 91.8 s | 1882 ns/day |
Trp-cage over 500 ps of implicit-solvent dynamics (100 frames, backbone aligned). Cartoon coloured N→C terminus. The mini-protein stays folded — thermal breathing, not unfolding.
Every run is fully reproducible — trajectories analyzed with MDTraj, figures generated from the raw output.
Same system (2661-atom TIP3P water box, PME, 3000 production steps), three OpenMM compute platforms. CUDA > OpenCL > CPU, as expected.